

Des recherches du qui pourraient conduire à de grandes avancées pour comprendre la maladie.
 Qu’est-ce que le syndrome de Cohen ?
Qu’est-ce que le syndrome de Cohen ?
Il s’agit d’une maladie génétique rare dont les signes cliniques sont très spécifiques chez les patients (dysmorphie faciale, microcéphalie, obésité tronculaire, hyperlaxicité ligamentaire, rétinopathie pigmentaire, neutropénie.)
Cette pathologie se transmet sur un mode autosomal récessif, c’est-à-dire qu’il faut que chacun des parents soit porteur d’une mutation sur les deux copies du gène d’intérêt. Si l’enfant hérite de l’allèle (gène d’intérêt) muté de son père et de sa mère, il sera malade.
Le diagnostic se pose souvent vers l’âge de 3 ans, notamment avec les retards d’acquisition constatés.
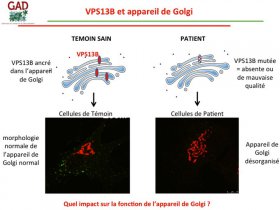
Le gène responsable de la maladie s’appelle le gène VPS13B.
Pour rappel, dans une cellule, il y a le noyau, les mitochondries et l’appareil de Golgi. Il a été montré en 2011 que VPS13B est une protéine qui appartient à la membrane de l’appareil de Golgi au sein de la cellule. Lorsque VPS13B est muté, la cellule a un appareil de Golgi totalement désorganisé.
Or, c’est dans l’appareil de Golgi que les protéines nouvellement synthétisées dans la cellule subissent des modifications post-traductionnelles comme les glycosylations.
La glycosylation est l’apposement de différentes sortes de sucres sur les protéines. C’est grâce à ce processus qu’on obtient une protéine fonctionnelle qui pourra faire son travail.
 Quelles sont les récentes découvertes dans la compréhension et le traitement de la maladie ?
Quelles sont les récentes découvertes dans la compréhension et le traitement de la maladie ?
Nous nous sommes posés plusieurs questions : un appareil de Golgi défragmenté peut-il être toujours fonctionnel ? Et si ce n’est pas le cas, les protéines seront-elles correctement glycosylées ?
Pour vérifier cela, nous avons analysé les protéines présentes dans le sérum des patients et avons découvert qu’il y avait un défaut majeur de glycosylation.
Nous avons réalisé des expériences de siRNA. Elles permettent de mimer sur des cellules normales en culture l’absence de VPS13B et de les rendre comme des cellules des patients.
On constate alors qu’on engendre un appareil de Golgi désorganisé et que les protéines issues de ces cellules présentent bien un défaut de glycosylation.
Plusieurs projets sont en cours au laboratoire pour comprendre pourquoi les patients atteints du syndrome de Cohen souffrent d’obésité tronculaire, de neutropénie ou encore de rétinopathie pigmentaire. Cette découverte du défaut de glycosylation apporte de nouvelles pistes de recherche.
Par cette découverte sur le défaut de glycosylation des protéines, nous imaginons que certaines protéines clés ne sont pas fonctionnelles. C’est sur la recherche de ces mécanismes que nous travaillons actuellement.
Des avancées thérapeutiques sont-elles envisageables ?
L’objectif est de comprendre l’implication du gène VPS13B dans les mécanismes qui conduisent à l’obésité, à la neutropénie et la rétinite pigmentaire.
Dans le projet sur l’obésité, nous avons constaté que les cellules des patients atteints du syndrome de Cohen sont capables d’accumuler plus de gras que des cellules normales. Ce fait corrèle avec l’obésité observée chez les patients. En parallèle, nous étudions les phénomènes de résistance à l’insuline de ces cellules, permettant probablement une prévention nutritionnelle auprès de ces patients afin d’éviter l’apparition d’un éventuel diabète.
En ce qui concerne la rétinopathie pigmentaire, l’objectif est de comprendre la dégénérescence de la rétine. Une cause possible est la mort des cellules par apoptose (mort programmée des cellules).
Si c’est le cas, nous définirons les étapes qui amènent à la mort des cellules et testerons différentes molécules pharmacologiques pour l’éviter ou la retarder. Ces projets devraient connaître des résultats dans les 2 ans à venir.
Une souris dont le gène Vps13b a été invalidé est en cours d’élaboration à l’institut clinique de la souris (ICS) à Strasbourg. Elle devrait présenter les symptômes de la maladie de Cohen et servir de modèle pour la compréhension des mécanismes responsables des symptômes de cette pathologie. Plusieurs molécules en vue de thérapie devraient être testées, notamment pour la rétine pigmentaire.
Mais cela prendra encore plusieurs années.
Le syndrome de Cohen : les signes cliniques
- Dysmorphie faciale : visages avec des traits spécifiques
- Microcéphalie : petit périmètre crânien entraînant une déficience intellectuelle
- Obésité tronculaire : Les membres sont d’apparence normale mais obésité au niveau du tronc. Extrémités effilées (très grands doigts).
- Hyperlaxicité ligamentaire : mouvements que les individus lambda ne sont pas capables de faire.
- Rétinopathie pigmentaire : la rétine se pigmente peu à peu et entraîne un fort handicap visuel (35% des patients sont aveugles ou fortement déficients visuels).
- Neutropénie : absence ou rareté de neutrophile dans le sang. Cellules capables de combattre les bactéries ou tout autre agent pathogène.